DeepMind puts the entire human proteome online, as folded by AlphaFold
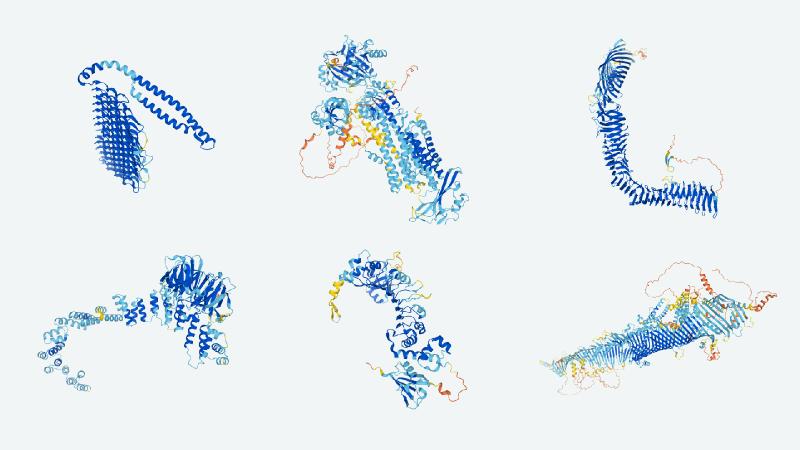

DeepMind and several research partners have released a database containing the 3D structures of nearly every protein in the human body, as computationally determined by the breakthrough protein folding system demonstrated last year, AlphaFold. The freely available database represents an enormous advance and convenience for scientists across hundreds of disciplines and domains, and may very well form the foundation of a new phase in biology and medicine.
The AlphaFold Protein Structure Database is a collaboration between DeepMind, the European Bioinformatics Institute and others, and consists of hundreds of thousands of protein sequences with their structures predicted by AlphaFold — and the plan is to add millions more to create a “protein almanac of the world.”
“We believe that this work represents the most significant contribution AI has made to advancing the state of scientific knowledge to date, and is a great example of the kind of benefits AI can bring to society,” said DeepMind founder and CEO Demis Hassabis.
From genome to proteome
If you’re not familiar with proteomics in general — and it’s quite natural if that’s the case — the best way to think about this is perhaps in terms of another major effort: that of sequencing the human genome. As you may recall from the late ’90s and early ’00s, this was a huge endeavor undertaken by a large group of scientists and organizations across the globe and over many years. The genome, finished at last, has been instrumental to the diagnosis and understanding of countless conditions, and in the development of drugs and treatments for them.
It was, however, just the beginning of the work in that field — like finishing all the edge pieces of a giant puzzle. And one of the next big projects everyone turned their eyes toward in those years was understanding the human proteome — which is to say all the proteins used by the human body and encoded into the genome.
The problem with the proteome is that it’s much, much more complex. Proteins, like DNA, are sequences of known molecules; in DNA these are the handful of familiar bases (adenine, guanine, etc.), but in proteins they are the 20 amino acids (each of which is coded by multiple bases in genes). This in itself creates a great deal more complexity, but it’s only the start. The sequences aren’t simply “code” but actually twist and fold into tiny molecular origami machines that accomplish all kinds of tasks within our body. It’s like going from binary code to a complex language that manifests objects in the real world.
Practically speaking this means that the proteome is made up of not just 20,000 sequences of hundreds of acids each, but that each one of those sequences has a physical structure and function. And one of the hardest parts of understanding them is figuring out what shape is made from a given sequence. This is generally done experimentally using something like x-ray crystallography, a long, complex process that may take months or longer to figure out a single protein — if you happen to have the best labs and techniques at your disposal. The structure can also be predicted computationally, though the process has never been good enough to actually rely on — until AlphaFold came along.
Taking a discipline by surprise
Without going into the whole history of computational proteomics (as much as I’d like to), we essentially went from distributed brute-force tactics 15 years ago — remember Folding@home? — to more honed processes in the last decade. Then AI-based approaches came on the scene, making a splash in 2019 when DeepMind’s AlphaFold leapfrogged every other system in the world — then made another jump in 2020, achieving accuracy levels high enough and reliable enough that it prompted some experts to declare the problem of turning an arbitrary sequence into a 3D structure solved.
I’m only compressing this long history into one paragraph because it was extensively covered at the time , but it’s hard to overstate how sudden and complete this advance was. This was a problem that stumped the best minds in the world for decades, and it went from “we maybe have an approach that kind of works, but extremely slowly and at great cost” to “accurate, reliable, and can be done with off the shelf computers” in the space of a year.
The specifics of DeepMind’s advances and how it achieved them I will leave to specialists in the fields of computational biology and proteomics, who will no doubt be picking apart and iterating on this work over the coming months and years. It’s the practical results that concern us today, as the company employed its time since the publication of AlphaFold 2 (the version shown in 2020) not just tweaking the model, but running it… on every single protein sequence they could get their hands on.
The result is that 98.5% of the human proteome is now “folded,” as they say, meaning there is a predicted structure that the AI model is confident enough (and importantly, we are confident enough in its confidence) represents the real thing. Oh, and they also folded the proteome for 20 other organisms, like yeast and E. coli, amounting to about 350,000 protein structures total. It’s by far — by orders of magnitude — the largest and best collection of this absolutely crucial information.
All that will be made available as a freely browsable database that any researcher can simply plug a sequence or protein name into and immediately be provided the 3D structure. The details of the process and database can be found in a paper published today in the journal Nature .
“The database as you’ll see it tomorrow, it’s a search bar, it’s almost like Google search for protein structures,” said Hassabis in an interview with TechCrunch. “You can view it in the 3D visualizer, zoom around it, interrogate the genetic sequence… and the nice thing about doing it with EMBL-EBI is it’s linked to all their other databases. So you can immediately go and see related genes, And it’s linked to all these other databases, you can see related genes, related in other organisms, other proteins that have related functions, and so on.”
[much more via the link ... no paywall]
Tags
Who is online

120 visitors
All that's left is for DeepMind to explain women to my ShallowMind ... or not, the mystery is too enthralling.
So artificial intelligence did all this?
Remember when the Terminator said he had detailed files on human anatomy?
All kidding aside, maybe this might be the breakthrough needed for diseases like diabetes
There's always the failed 'scientists' in basement protein soup kitchens to worry about.
AI is a foundation for prescient digital life extention.